About Sanger sequencing
How can the Sequencing and Bioinformatics Consortium help me with Sanger sequencing?
We provide automated DNA sequencing using Applied Biosystems 3730XL instrumentation and chemistries. This instrument is the gold standard for high-throughput genetic analysis, allowing up to 96 samples to be sequenced in a single run.
What do I need to know about submitting samples?
How do I access dnaLIMS?
You can access our online DNA submission portal here.
You will need to register for an account by clicking on “Create Login Account for dnaLIMS”, and filling in your login and contact information as directed. Once your account is created, you will be able to enter your order and sample information.

How do I know that my samples have been handled appropriately, the sequencing process worked, and that the data I received is from my sample?
How do I assess the quality of my sequence?
How do I know if my sequence is of good quality?
Following sequencing, we inspect each sequence prior to data release. Through our analysis software, our technicians evaluate:
- The sequences’ overall sample scores to ensure that the sequence has reliable and usable data.
- The length of the sequence to determine if the sequence generated is longer or shorter than expected.
If any issues are identified in your sequencing results, we will notify you of the issues when we send out your data.
As a general rule, a good quality sequence should have singular peaks that are well defined, sharp and have even spacing between them. The peak height should be high enough that the background noise is minimal in comparison. A sequence failure will often display some of the following characteristics:
- No signal is produced and no peaks are identified.
- Base calls are replaced with N’s.
- All noise in the electropherogram
- The low peak signal in conjunction with baseline noise renders the sample’s signal indecipherable

Figure 1: The sequencing software failed to call any bases with background noise interfering with sample peaks
Why do I need to review the chromatogram file and not just the text file?
Reviewing your chromatogram is an essential part of analyzing and troubleshooting your Sanger sequencing results. As an example, review the text file and chromatogram below. If you only reviewed your data in a text file and blasting this confirms what you were expecting (without ever looking at your chromatogram), you would likely be pleased with your sequencing results. However, your data analysis would be incomplete as a quick view of the overlapping peaks in the chromatogram would suggest your results are not as certain as the sequence initially suggests. As such, your chromatogram acts as a visual control. The Sequencing team will notify you by email if we identified any issues in your sequencing results, which would be apparent in the chromatogram. If your Sanger sequencing result is of lower quality, as shown in the left image of Figure 2, sequencing should be repeated. Please review our Resequencing Policy for more information.

Figure 2: (Left) Text file and corresponding chromatogram trace showing poor quality sequencing data due to spectral pull-up, (Right) Text file and corresponding chromatogram trace showing good quality sequencing data
How do you tell the difference between traces of a PCR product and a plasmid?
Several DNA polymerases such as Taq add a single nucleotide base to the 3‘ end of amplified DNA fragments. The AmpliTaq DNA Polymerase we use during PCR amplification in our Sanger sequencing adds a 3’ adenine overhang to the end of the PCR product. As such, sequenced PCR products are easily identified by their signature adenine peak at the end of the amplicon sequence (Figure 3). This adenine peak is generally larger than its neighbouring peaks and should be well defined, appearing at the end of your sequenced amplicon product (only visible if the amplicon is shorter than the length of sequencing read).

Figure 3: Chromatogram showing the A-tail seen at the end of an amplicon generated with AmpliTaq.
When sequencing a plasmid, no A-tail or abrupt stop is seen at the end of the sequencing product. In a successfully sequenced plasmid, the bases called may seemingly never end (Figure 4). The sequencing software however will stop calling bases once the limits of the 3730XL DNA sequencing instrument are reached. Similarly, longer PCR amplicons, such as those exceeding 1kb, may exhibit comparable characteristics to those seen in a plasmid trace.

Figure 4: Raw trace file for a successfully sequenced plasmid
My results aren’t what I expected, can you help me understand what went wrong?
What are the effects of too much or too little DNA in Sanger sequencing?
A ‘ski slope’ effect is often seen in samples that:
- are over- or underloaded with DNA
- use insufficient or too much primer for the sequencing reaction
- use degraded template
As such, it is essential that template material submitted for Sanger sequencing is correctly quantified. We will adjust the amount of DNA input into the reaction, based on the quantifications provided on your sample submission form, as appropriate for the template type and length. Refer to "How should I quantify my DNA prior to submission?" for recommendations on how to quantify your DNA prior to submission for Sanger sequencing.

Figure 5: Ski slope effect
Why are there multiple overlapping peaks at the same position? Does this mean my data is unusable?
Overlapping peaks at the same position is often referred to as a ‘mixed trace’ or ‘peaks on peaks’. We will notify you if multiple overlapping peaks are observed in your sequence. Whether or not you can use the data will depend on the goals of your sequencing.
Common explanations for mixed traces are listed below. If unexpected mixed traces occur, we suggest double checking your primers for specificity. In certain cases, the primers may need to be redesigned. Examining your template using agarose gel electrophoresis prior to submission for Sanger sequencing is also recommended, as it will allow you to identify the presence of multiple templates
- Mutations such as insertions or deletions in some of the DNA fragments.
- Multiple primer binding sites on the template.
- Multiple templates or primers present in the reaction.

Figure 5: Example of a mixed trace
What are dye blobs, and what are they caused by? Does this mean the data is unusable?
Dye blobs are sharp peaks seen in the chromatogram that are a result of unincorporated dye terminators that were not fully eliminated during the cleanup of the sequencing reaction. At the SBC, we purify after the sequencing reaction to remove unincorporated dye terminators, enzymes and salts that remain following the cycling reaction.
There are a few cases where dye blobs may persist, even following the purification process. For example, samples that failed during the sequencing reaction resulting in weak or no signal may leave a greater proportion of unincorporated dye terminators. This is commonly seen in our non-template controls, where the lack of amplifiable template results in weak signals and a large dye blob. Additionally, the presence of any contaminants such as salt or EDTA in your storage buffers may prevent thorough amplification of the template, resulting in dye blobs.
Dye blobs generally occur in the 70 bp region. In more extreme cases, they may also occur in the 120 bp region. If dye blobs are observed in these regions, we will notify the client about their presence. Even though dye blobs may be observed in your sample, the underlying peaks should allow you to identify your bases manually. However, please contact us at sanger.submissions@ubc.ca if this is not the case.

Figure 6: Dye blob occurring in the 70bp region
What are C and G shoulders?
C or G shoulders are described as irregular C or G peaks seen in a chromatogram sequence following sequencing. The end of the C or G peak is not sharp and well defined as seen in a good peak but instead shows an irregularity at the end of the peak where the peak trace extends into a “shoulder”. Please contact us at sanger.submissions@ubc.ca for more information if C or G shoulders are interfering with your data quality.

Figure 7: Chromatogram showing good quality sequence, absent of C or G shoulders
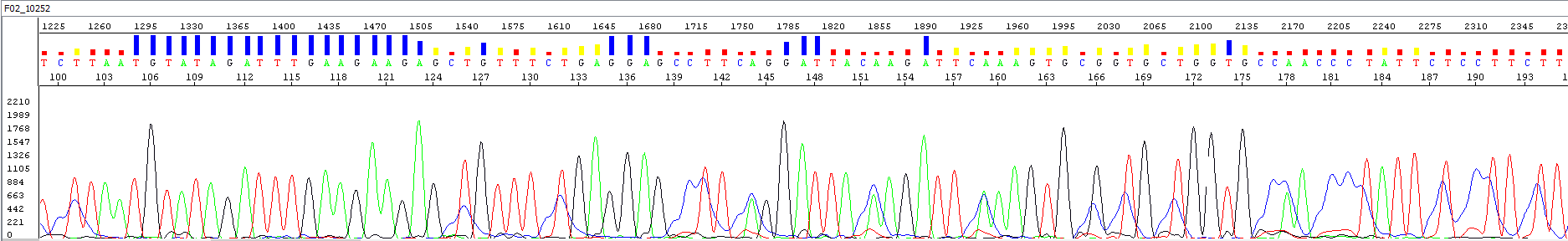
Figure 8: Chromatogram showing the presence of C shoulders
I observed a loss of resolution near the end of my sequence ~800bp. Why does this happen?
As the sequence approaches the resolution limits of the 3730XL sequencer, the probability of errors in base calling increases (Ewing & Green, 1998). Towards the ends of the sequence, the system sometimes has difficulty resolving fragments, which results in lower quality base calling as the base peaks are not well resolved and bleed into each other.
You may notice the following in lower quality regions:
- a reduced quality score
- broader overlapping peaks
- the presence of peaks with no bases called by the sequencing software
Figure 9: Example of high quality, well-defined peaks

Figure 10: Reduction of quality around the 800th base

Figure 11: Low-quality peaks near the end of the sequence
Why are the first 15-40 nucleotides in my sequence undeterminable and of poor quality?
The first 15 to 40 nucleotides of a sequence are often poor in quality. The poor quality is due to the Sanger sequencing technology itself, as the technology involves the use of bulky ‘big’ dyes bound to terminator bases. Although these dyes allow the instrument to differentiate between nucleotides during sequencing, the large molecular weights and charges on these dyes cause a shift in migration for the sequencing reaction products undergoing electrophoresis. At shorter fragment lengths, such as those seen in the first 40 nucleotides, these shifts will be more dramatic as the dye composes a greater percentage of the total molecular weight and charge of the sequencing product. Additionally, the presence of unincorporated dyes or incomplete removal of primer dimers following PCR purification may contribute to the messy peaks seen at the beginning of the sequence.
If you require high-quality bases in the first 40 nucleotides of your sequence, we suggest redesigning your primer further upstream of your sequence of interest to avoid any reduction in sequence quality.

Figure 12: Low-quality sequence in the first 40 bases
Resequencing Policy
References
Choi, J. S., Kim, J. S., Joe, C. O., Kim, S., Ha, K. S., & Park, Y. M. 1999. Improved cycle sequencing of GC-rich DNA template. 1999. Experimental Molecular Medicine, 31, 31(1) 20-24. DOI: 10.1038/emm.1999.3
Ewing, B., & Green, P. 1998. Base-Calling of Automated Sequencer Traces Using Phred II Error Probabilities. Genome Research, 8, 186-194. Cold Spring Harbor Laboratory Press. ISSN 1054-9803-98
Shapter, F. M., & Waters, D. L. 2014. Genome Walking. Methods Molecular Biology, 1099, 133-146. DOI: 10.1007/978-1-62703-715-0_12.